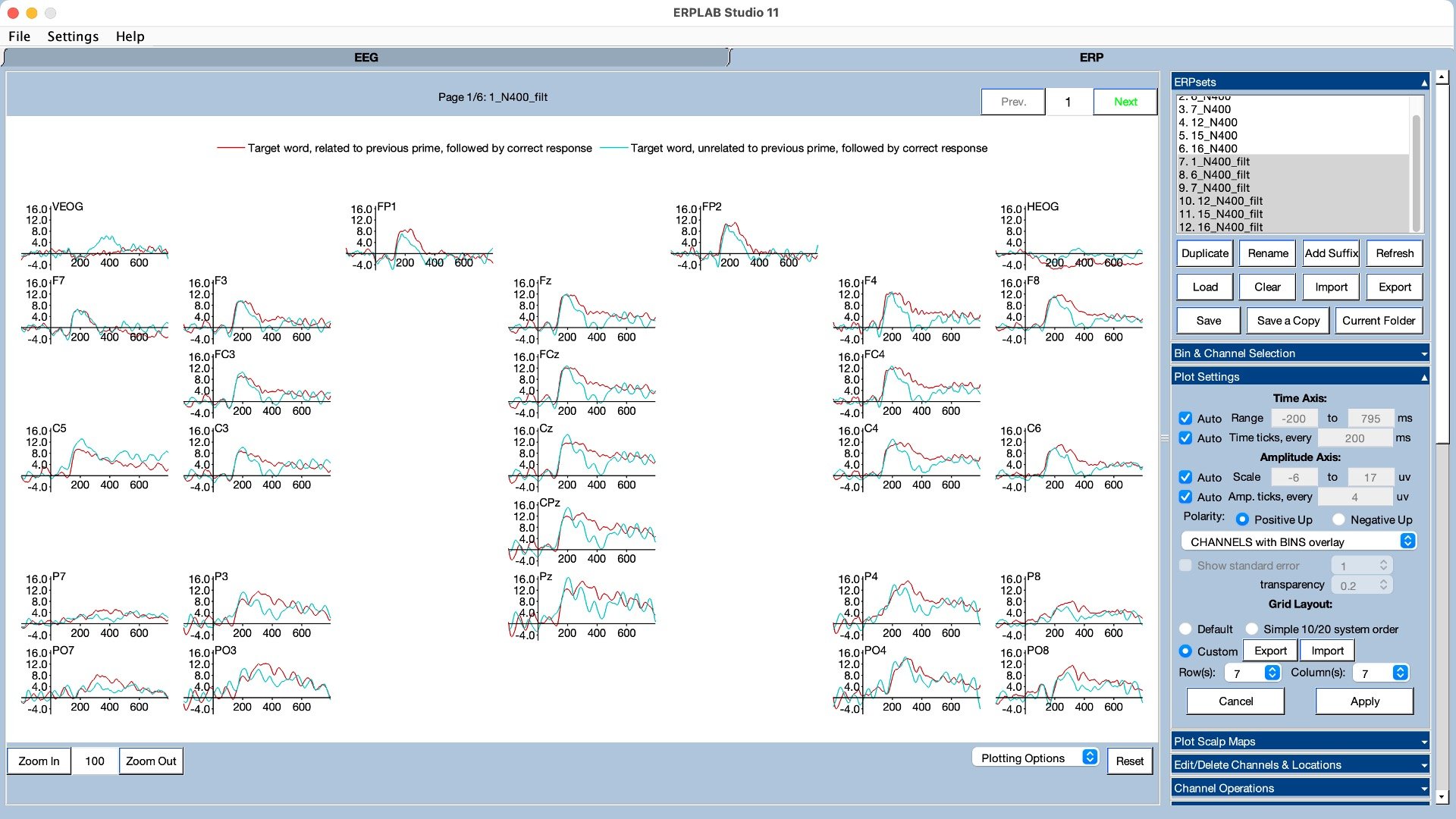
We also used ERP decoding to test whether the exact location of the dot was better stored in working memory on oddball trials than on standard trials (click here for information about how ERP decoding works and how you can decode your own data using ERPLAB Toolbox). As shown in Figure 5, we could decode the location of the dot better on oddball trials than on standard trials during the period following the P3b component. Note, however, that this was a pretty small difference that only barely crossed the threshold for statistical significance (p = .048). I would really like to see this effect replicated before fully believing it. However, the behavioral effect was rock solid (and replicated in a follow-up experiment).
What can we conclude from these findings? When we started the project, I knew that it would be difficult to draw any strong causal conclusions about the relationship between the P3b component and working memory updating. That is, even if we saw both a larger P3b and improved working memory on oddball trials, this would just be a correlation and could potentially be explained by a third variable such as attention. But if we saw a big difference in working memory between oddballs and standards, and if we found that working memory was better on trials with larger P3b amplitudes, this would be at least consistent with the idea that the process that produces the P3b on the scalp is also involved in working memory encoding.
However, although we saw an enormous difference in P3b amplitude between oddball trials and standard trials, we saw only small differences in working memory between oddballs and standards, whether measured via behavioral response errors on probe trials or EEG decoding accuracy. If the process that generates the scalp P3b voltage plays a major role in working memory encoding, then we would have expected a much larger working memory difference between oddballs and standards. Moreover, although we found that participants with larger P3b amplitudes had smaller response errors, we did not find any evidence that working memory was any better on trials with larger P3b amplitudes (even though we looked very hard for such a relationship). The bottom line is that, although I was really hoping we would finally provide some direct evidence for the working memory encoding hypothesis, the results of this study have actually convinced me that the P3b is probably not related to working memory encoding.
What, then, explains the small but statistically significant differences in working memory accuracy between oddballs and standards, along with the subjectwise correlation between P3b amplitude and behavioral response errors? A very plausible explanation is that both the P3b component and working memory encoding are facilitated by increased attention. That is, there are several sources of evidence that rare events trigger increased attention, and this could independently produce a larger P3b and improved working memory.
Of course, this is just one experiment, so I wouldn’t say that the working memory encoding hypothesis is completely dead. But given our new findings and the general lack of direct evidence for the hypothesis, it’s on life support.
If the P3b doesn’t reflect working memory encoding, then what does it reflect? This seems like a significant question: the P3b is huge and is observed across a broad range of experimental paradigms, and it’s reasonable to assume that the underlying process must be important for the brain to devote so many watts of energy to it. In fact, I find it embarrassing that our field has not answered this question in the 60 years since the P3b was first discovered.
My best bet is that the P3b is related to the process of making decisions about actions (where the term actions is broadly construed to include the withholding of responses and mental actions such as counting). This is related to the fact that the amplitude of the P3b is related to the probability of a task-defined category, not the probability of a physical stimulus category. Rolf Verleger has a nice review of the evidence for this idea (Verleger, 2020). But it is still not clear to me why the brain devotes so many watts of energy to creating a large P3b when a rare task-defined category occurs. Verleger notes that several hypotheses about the P3b are compatible with the finding of a larger P3b for oddballs than for standards, but in my view these hypotheses have a hard time explaining the enormous size of the P3b observed for oddballs. This is a longstanding mystery in need of a solution!